Синдром на Блум: Симптоми, причини и лечение
Синдромът на Блум (BS) е рядко заболяване на автозомно рецесивно наследяване, което се характеризира главно с три аспекта: забавяне на растежа, свръхчувствителност към слънцето и телеангиектазия в лицето (разширяване на капилярните съдове). Тези пациенти имат геномна нестабилност, която ги предразполага да развиват лесно рак.
Той е открит от дерматолога Дейвид Блум през 1954 г. чрез наблюдение на няколко пациенти, които са представили джуджето и телангиоктатната еритема (зачервена кожа, дължаща се на дилатация на кръвоносните капиляри) (Elbendary, 2015).
Този синдром може да се нарече и вроден телеангиоктатен еритем или синдром на Bloom-Torre-Machacek.
Причини за възникване на синдрома на Блум
Синдромът на Блум е автозомно рецесивно заболяване, т.е. мутация трябва да се появи и в двата алела на BLM гена, както от майката, така и от бащата (Ellis et al., 1995). Родителите не е задължително да имат това заболяване, но могат да бъдат носители на мутиралия ген, без да имат симптоми.
Повече от 60 мутации са открити в BLM гена в синдрома на Bloom, като най-често е заличаването на 6 нуклеотида в позиция 2281 и заместването с 7 други (Elbendary, 2015).
Според Genetics Home Reference (2016) генът BLM е отговорен за изпращане на инструкции за създаването на протеин RecQ, който е част от семейството на хеликази.
Това, което правят хеликазите, е да се присъединят към ДНК и временно да се отделят двете нишки от него, които обикновено са свързани в спирала, с цел да се развият процеси като репликация (или копиране на ДНК), подготовка за клетъчно делене и ремонт. на увреждане на ДНК.
Накратко, RecQ хеликазите са важни за поддържане на структурата на ДНК и затова са известни като "геномни грижи".
Например, когато една клетка ще се раздели, за да образува две нови клетки, ДНК, която е в хромозомите, трябва да бъде копирана, така че всяка нова клетка да има две копия на всяка хромозома: един от бащата и друг от майката.
ДНК, копирана от всяка хромозома, има две идентични структури, които се наричат сестрински хроматиди, и те са свързани в началото, преди да се раздели клетките.
На този етап те обменят части от ДНК между тях; това, което е известно като обмен на сестрински хроматиди. Изглежда, че този процес е променен в болестта на Блум, тъй като BLM протеинът е повреден и това контролира правилния обмен между сестринските хроматиди и че ДНК остава стабилна по време на копирането.
Всъщност има средно 10 повече от нормалния обмен между хроматидите в синдрома на Блум (Seki et al., 2006).
От друга страна, има и прекъсвания в генетичния материал при това заболяване, които причиняват влошаване на нормалните клетъчни дейности, които поради липсата на BLM протеин не могат да бъдат поправени.
В действителност, някои експерти класифицират този синдром като "синдром на хромозомния пробив", тъй като той е свързан с голям брой прекъсвания и пренареждания на хромозомите.
Тази нестабилност на хромозомите води до по-голяма вероятност от развитие на заболявания. Например, поради липсата на BLM протеин, те не могат да се възстановят от увреждане на ДНК, което може да причини ултравиолетова светлина и следователно тези пациенти са фоточувствителни.
Освен това, засегнатите имат имунен дефицит, който ги прави по-податливи на инфекция.
От друга страна, те имат голяма вероятност да развият рак във всеки орган от неконтролираното разделяне на клетките, което се проявява главно левкемия (това е вид рак на кръвта, характеризиращ се с излишък на бели кръвни клетки) и лимфом (рак в лимфния възел на системата). имунната).
Открити са и неуспехи в действието на гена FANCM, който е отговорен за кодирането на протеини ММ1 и ММ2, които също служат за възстановяване на увреждане на ДНК.
Това са тези, които са свързани както с този синдром, така и с анемията на Фанкони. Ето защо виждаме, че тези две болести са сходни по своя фенотип и с предразположеност към хематологични тумори и недостатъчност в костния мозък.
Както и да е, молекулярните механизми, които засягат хромозомите в синдрома на Блум, са все още в процес на проучване.
Какво е неговото разпространение?
Синдромът на Блум е сравнително рядък, известни са само около 300 случая, описани в медицинската литература. Въпреки че това заболяване се среща в много етнически групи, изглежда, че е много по-често при евреите от Ашкенази, което представлява 25% от пациентите с този синдром.
Всъщност, в рамките на тази етническа група честотата на представяне на синдрома може да достигне 1%. Също така е установено, макар и по-рядко, в японски семейства.
Що се отнася до секса, мъжете изглежда са малко по-склонни да имат заболяването, отколкото жените, като съотношението е 1, 3 мъже за 1 жена.
Какви са нейните симптоми?
Това състояние вече се случва през първите месеци от живота и за сега никой от пациентите не е живял повече от 50 години.
- Злокачествени тумори : причинени от геномна нестабилност, както е обяснено по-горе, са основната причина за смърт при засегнатите от този синдром. Според Националната организация за редки заболявания (2014) около 20% от засегнатите от синдрома на Блум ще развият рак. При тези пациенти рискът от развитие на рак е между 150 и 300 пъти по-голям, отколкото при хората без това заболяване.
- Имунодефицит, който варира по тежест според пациента и предразполага към различни инфекции. Това се дължи на дефицит в пролиферацията на лимфоцити (бели кръвни клетки), проблеми в синтеза на имуноглобулин (антитела на имунната система) и нисък отговор на стимулация от митогени (които контролират деленето и растежа на клетките).
- Дефектите в Т и В лимфоцитите са чести, засягащи развитието на имунната система.
- Неправилното функциониране на имунната система може да доведе до инфекция на ушите (предимно отит), пневмония или други признаци като диария и повръщане.
- Фоточувствителност : това е прекомерна чувствителност на ДНК към ултравиолетовите лъчи, която се поврежда. Счита се за форма на фототоксичност или клетъчна смърт, която уврежда кожата на засегнатите, когато слънцето се удари.
- Намаляване на плодовитостта или безплодието . Всъщност при мъжете има невъзможност да се изчака. При жените има много ранна менопауза.
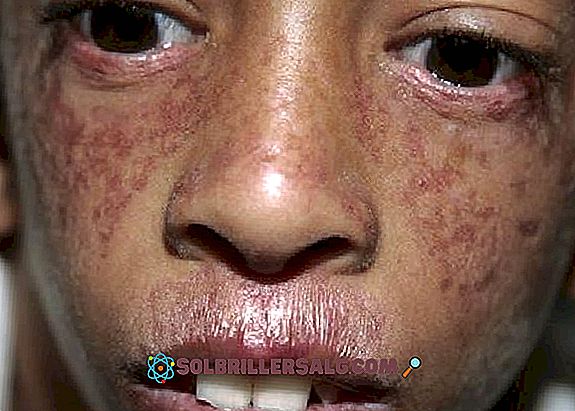
- Кожни прояви : освен фоточувствителност, се появява и poikiloderma, афектация на кожата, която се появява главно в областта на шията, появяващи се хипопигментирани зони, други хиперпигментирани области, телеангиектазии и атрофия. Често се наблюдават червени петна по кожата, които са свързани с излагане на слънце (особено на лицето).
Друг наблюдаван кожен проблем е телеангиектазията, която се разглежда като червеникави изригвания по лицето, причинени от дилатация на малки кръвоносни съдове. Той се появява като модел на "пеперуда", покриващ носа и бузите.
- Анормални кафяви или сиви петна могат да се появят и на други части на тялото (петна „café au lait“).
- Забавяне в развитието, което вече се проявява при бебета. Малките обикновено имат отличителна глава и лице, по-тесни и по-малки от нормалните.
- Около 10% от засегнатите в крайна сметка развиват диабет (Национална организация за редки заболявания, 2014).
- Много остър глас .
- Промени в зъбите .
- Аномалии в очите, ушите (изпъкнали уши), ръце или крака (като полидактилия, което се появява, когато пациентът има повече пръсти от нормалното).
- Пилонидални кисти.
- Проблеми с храненето : те са забелязани особено при бебета и малки деца, показващи липса на интерес към храненето. Той е съпътстван многократно от тежък гастроезофагеален рефлукс.
- Интелектуалните способности са променливи, така че при някои пациенти те са по-влошени, а в други са в нормално състояние.
Как се диагностицира?
Може да се диагностицира чрез някой от следните тестове:
- Цитогенетични тестове, които измерват хромозомните аберации и нивото на обмен на сестрински хроматиди.
Възможно е да се наблюдава наличието на четири-радиални асоциации (обмен на четирихватни хроматиди) в лимфоцити, култивирани в кръвта, за да се провери дали има високи нива на обмен на сестрински хроматиди във всяка клетка, хроматични пропуски, счупвания или пренареждания; Или вижте директно, ако има мутации в BLM гена.
Тези тестове могат да открият здрав индивид, който носи мутации в BLM гена и може да ги предава на тяхното потомство.
Агенцията по храните и лекарствата на САЩ (FDA) обяви през февруари 2015 г. комерсиализацията на генетичен тест от "23andMe", който може да бъде полезен за откриване на ранно присъствие на това заболяване.
Наличието на този синдром трябва да се подозира, ако съществуват такива клинични състояния:
- Забавяне на значителния растеж, наблюдаван от вътрематочния период.
- Наличие на еритеми върху кожата на лицето след излагане на слънце.
Не бъркайте с ...
Следните синдроми трябва да бъдат взети под внимание, за да бъдат изключени, преди да се диагностицира синдромът на Блум:
- Други синдроми на автозомно-рецесивна хромозомна нестабилност, които са свързани с прекъсвания и пренареждане на хромозоми, което прави субекта особено уязвим към някои видове рак, като например: анемия на Fanconi, атаксия телеангиектазия или xeroderma pigmentosa, които включват други гени, а не BLM,
- Синдром на Cockayne, който се състои от наследствено заболяване, проявяващо се със забавено развитие, фоточувствителност и стареене в ранна възраст. Това е рядка форма на джудже.
- Синдром на Ротмунд-Томсън : изключително рядко срещано и проявяващо се с типични кожни аномалии, дефекти на косата, ювенилни катаракти, къс ръст и скелетни промени, като краниофациални малформации. Прилича на синдрома на Блум при възпаление на кожата, пойкилодермия, дегенерация на кожата (атрофия) и телеангиектазия.
лечение
Няма специфично лечение за синдром на Блум, т.е. за прекомерния брой мутации. Напротив, интервенциите са насочени към облекчаване на симптомите, предлагане на подкрепа и предотвратяване на усложнения.
- Опитайте се да не се излагате директно под слънцето.
- Използвайте подходящ слънцезащитен крем.
- Проследяване от дерматолог, за лечение на петна, зачервяване и възпаление на кожата.
- Използвайте антибиотици за инфекции.
- Периодични медицински прегледи за откриване на възможни случаи на рак, главно когато тези пациенти достигнат зряла възраст. Трябва да се опитаме да бъдем внимателни към възможните симптоми, тъй като има тумори, които изискват ранно хирургично отстраняване за тяхното възстановяване. Някои методи за ранна диагностика на рак са мамография, Papanicolaou или вагинална цитология или колоноскопия.
- Контролирайте, че тези деца получават необходимите хранителни вещества, които се опитват да се намесят в храносмилателния рефлукс. За тази цел в горната част на чревния тракт може да се постави тръба за допълващо хранене, докато спите. Това може да увеличи малко мастните натрупвания, но изглежда не оказва влияние върху самия растеж.
- Проучете съществуването на диабет, за да го лекувате възможно най-скоро.
- Ако индивидът има рак, може да се предвиди трансплантация на костен мозък.
- Семейни и други групи за подкрепа и асоциации със сходни болести, така че засегнатото лице да се развива като човек, с възможно най-високо качество на живот.
- Ако семейството е имало случаи на това заболяване или от семейството на съпруга, генетичното консултиране би било полезно за получаване на информация за естеството, наследството и последствията от този тип нарушения, за да допринесе за вземането на медицински решения и лично.







